A esclerose múltipla (EM), a mais comum doença desmielinizante crônica do sistema nervoso central (SNC) e a causa mais comum de incapacidade não traumática em adultos jovens, acomete cerca de 2,5 milhões de pessoas no mundo todo.1,2 No Brasil, dados publicados indicam ampla variação da prevalência por região geográfica, com taxas entre 1,3 e 27,2 casos por 100 mil habitantes.3,4
Embora certos achados sejam típicos da EM, suas manifestações podem variar amplamente de um paciente para outro. O processo inflamatório de base, caracterizado por áreas de desmielinização imunomediada no cérebro e na medula espinhal, é influenciado por fatores genéticos e ambientais. A lesão da bainha de mielina (cobertura protetora composta de lipídios que isola os nervos e transmite os impulsos elétricos ao longo do axônio) resulta em placas ou lesões que levam aos sintomas clínicos, com disseminações temporal e espacial. As manifestações mais comuns incluem distúrbios sensitivos nas extremidades, disfunção do nervo óptico e do trato piramidal, comprometimento das funções vesicais, intestinais e sexuais, depressão, ataxia e diplopia, entre outras. Na maioria dos casos, a EM segue uma evolução recorrente-remitente, com episódios breves e recorrentes de déficits neurológicos. Nos intervalos entre esses surtos, os sintomas podem desaparecer completa ou parcialmente. Entretanto, os déficits neurológicos frequentemente tornam-se permanentes à medida que a doença avança.1,2
Os diferentes fenótipos da esclerose múltipla
– A síndrome clinicamente isolada (CIS, do original clinically isolated syndrome) constitui o primeiro evento desmielinizante típico da EM e pode consistir em um episódio de neurite óptica (NO), mielite transversa (MT) ou outras manifestações comuns, com achado correlato em exame de neuroimagem. Pacientes com CIS não têm história de episódios prévios de desmielinização. O desenvolvimento dos sintomas é agudo ou subagudo e a duração mínima é de 24 horas, com ou sem recuperação. É importante que ocorram na ausência de febre ou de infecção. Pode ser monofocal ou multifocal.5
– A EM recorrente-remitente (EMRR) é o fenótipo mais comum da doença e corresponde a cerca de 85% a 90% dos casos iniciais. A EMRR é caracterizada por uma série de surtos clínicos com plena recuperação ou déficits residuais. Os sintomas podem durar de alguns dias a vários meses, com mínima progressão da doença entre os surtos. Quando não tratados, até 50% dos pacientes progridem para a fase secundariamente progressiva da EM dentro de 10 anos.5 (Figura 1)
– Na EM secundariamente progressiva (EMSP), o paciente apresenta piora neurológica gradual insidiosa, com ou sem surtos ocasionais, remissões parciais e platôs. Não há critérios estabelecidos para determinar quando a EMRR se converte em EMSP, mas o diagnóstico de EMSP é feito retrospectivamente, com base na história clínica. Na maioria dos pacientes, a transição da EMRR para a EMSP ocorre dentro de 10 a 20 anos após o início dos sintomas.5 (Figura 1)
– A EM primariamente progressiva (EMPP) é o fenótipo clínico menos comum de EM. Caracteriza-se por deterioração neurológica progressiva e acúmulo de incapacidade desde o início dos sintomas, sem recorrências, nem platôs, nem melhoras temporárias. (Figura 1) Os achados clínicos que distinguem a EMPP dos outros fenótipos da doença incluem paraparesia espástica assimétrica, predileção pelo envolvimento medular ao exame de ressonância magnética nuclear (RMN) e ausência de surtos de recorrência. O diagnóstico é feito exclusivamente com base na história clínica.5
Figura 1. Padrões de evolução da incapacidade cumulativa nos diferentes fenótipos de EM
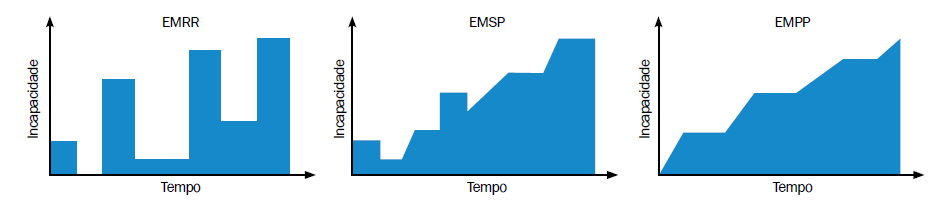
Os diferentes “tipos” de EM não constituem, entretanto, entidades independentes entre si, mas ocorrem ao longo de um espectro contínuo de atividade da doença – com lesões inflamatórias focais em uma extremidade e neurodegeneração progressiva na outra. As lesões inflamatórias focais constituem a característica típica da EMRR. Tanto na EMSP como na EMPP, a incapacidade cumulativa não é bem explicada pelas lesões observadas à RMN e ocorre independentemente dos surtos de recorrência. As formas progressivas de EM são mais comumente associadas a uma apresentação motora e a pior prognóstico, com acúmulo mais rápido de incapacidades. As formas progressivas também são mais difíceis de tratar e as chama- das drogas modificadoras da doença (DMDs) são geralmente menos efetivas em alterar a evolução desses fenótipos da doença.5
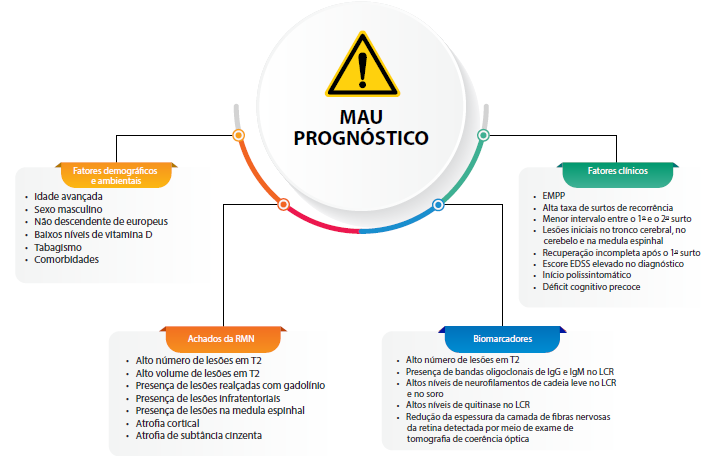
Fatores de mau prognóstico
Alguns aspectos clínicos da EM estão associados à extensão da incapacidade em longo prazo e, portanto, têm valor prognóstico. Em um dos maiores estudos longitudinais de prognóstico da EM, que incluiu 28 mil pacientes-anos de acompanhamento, a frequência de surtos de recorrência nos primeiros 2 anos e o tempo entre o primeiro e o segundo surto da doença foram importantes fatores preditivos dos desfechos de incapacidade em longo prazo. A figura 2 lista esses e outros fatores demográficos e ambientais, clínicos, bem como achados da RMN e biomarcadores associados a pior prognóstico em pacientes com EM.6
Figura 2: Fatores de mau prognóstico
Diagnóstico da EM – Os critérios de McDonald
O diagnóstico de EM é feito por meio de uma combinação que abrange história clínica, exame neurológico, RMN e exclusão de outros recursos diagnósticos. Outros exames auxiliares, como análises do líquido cefalorraquidiano (LCR), potenciais evocados e tomografia de coerência óptica, podem ser úteis, mas geralmente não são necessários.2
Os critérios de McDonald, inicialmente propostos em 2001 por um painel internacional de especialistas para o diagnóstico da EM,7 foram revisados em 2005,8 2010,9 201610 (com contribuições do grupo MAGNIMS para critérios referentes à RMN) e 201711,12 (última revisão publicada; Quadro 1).
Quadro 1. Critérios de McDonald de 2017 revisados e adaptados11,12
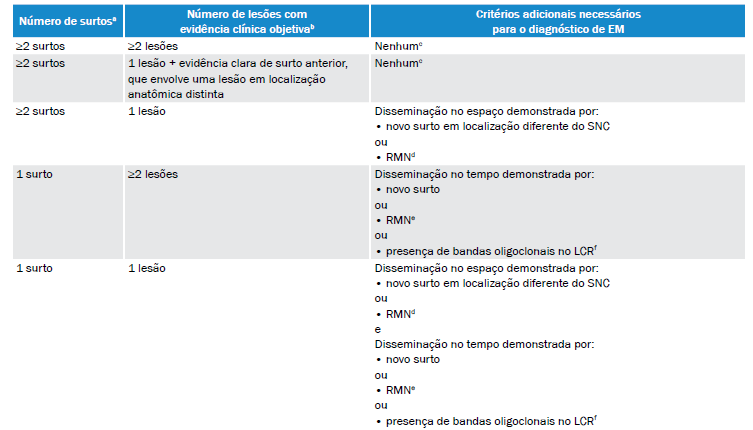
(a) Definição de surto: todo evento reportado pelo paciente, ou objetivamente observado, que seja típico de um episódio inflamatório desmielinizante agudo e tenha duração de pelo menos 24 horas, na ausência de infecção ou febre. O evento deve ser documentado por meio de exame neurológico realizado durante sua manifestação clínica. Alguns eventos históricos para os quais não haja achado neurológico documentado, mas que sejam típicos de EM, podem prover evidência suficiente de um evento desmielinizante prévio. Relatos de sintomas paroxísticos (históricos ou correntes) devem, no entanto, consistir em múltiplos episódios com duração superior a 24 horas. (b) O diagnóstico baseado em evidência clínica objetiva de duas lesões é o mais seguro. A evidência histórica de surto prévio, na ausência de achados neurológicos objetivamente documentados, pode incluir episódios com sintomas e evolução característicos de um evento desmielinizante inflamatório prévio. Pelo menos um surto, entretanto, deve ter seu suporte em achados objetivos. Na ausência de evidência objetiva residual, é necessário ter cautela. (c) De acordo com os critérios de McDonald originais (revisão de 2017),9 não são necessários testes adicionais, entretanto o Protocolo Clínico e Diretrizes Terapêuticas (PCDT) de EM10 considera que qualquer diagnóstico de EM deve ser realizado com acesso à neuroimagem. (d) Ressonância magnética para DIS (≥1 lesões hiperintensas em T2 na RM, sintomáticas ou assintomáticas, características de EM, em ≥2 das seguintes áreas do sistema nervoso central: periventricular, cortical/justacortical, infratentorial e da medula espinhal). (e) Ressonância magnética para DIT (presença simultânea de lesões captantes e não captantes de gadolínio em qualquer exame de RM, ou nova lesão hiperintensa em T2 e/ou captante de gadolínio, quando comparada a uma RM prévia, independentemente do momento em que foi realizada). (f) A presença de bandas oligoclonais no LCR não demonstra DIT, contudo pode substituir essa demonstração.
Adaptado de: Thompson AJ, et al. Lancet Neurol. 2018 Feb;17(2):162-73;12 Ministério da Saúde. PCDT de EM. 2024 [acesso em 29 out. 2024]. Disponível em: https://www.gov.br/conitec/pt-br/midias/ protocolos/pcdt-de-esclerose-multipla.12
No congresso anual do Comitê Europeu de Tratamento e Pesquisa da Esclerose Múltipla (ECTRIMS, do original European Committee for Treatment and Research in Multiple Sclerosis), realizado em setembro de 2024, foi proposta nova revisão dos critérios de McDonald, que ainda aguarda publicação. Entre as principais alterações propostas está a inclusão de lesões do nervo óptico como uma “quinta topografia” para demonstração da disseminação no espaço (DIS) da EM.13
O valor da RMN para o diagnóstico, prognóstico e monitoramento de pacientes com EM já está bem estabelecido e sua implantação tem sido especificada, com discretas variações, em diversos consensos e diretrizes internacionais dos Estados Unidos e da Europa, entre outros. Embora represente um grande desafio diante das diferenças entre os sistemas de saúde e as práticas clínicas dos países, essas diretrizes têm orientado neurorradiologistas e neurologistas a padronizar os protocolos de aquisição de imagens e a cronologia dos exames, de modo a incentivar as sociedades nacionais e internacionais a estabelecerem recomendações similares.14
No Brasil, o Protocolo Clínico e Diretrizes Terapêuticas (PCDT),12 documento elaborado pela Comissão Nacional de Incorporação de Tecnologias no Sistema Único de Saúde (CONITEC) para orientar o manejo da EM no âmbito do Sistema Único de Saúde (SUS), estratifica a atividade das formas progressivas (EMSP e EMPP) e recorrente (EMRR) da EM em alta, moderada ou baixa, com base em achados da RMN:
- EM de baixa ou moderada atividade: indícios de atividade da doença, sem preencher critérios para alta atividade.12
- EM de alta atividade:
1. PACIENTES VIRGENS DE TRATAMENTO: ≥2 surtos e ≥1 lesão captante de gadolínio ou aumento de pelo menos duas lesões em T2 no ano prévio;12
2. PACIENTES PREVIAMENTE TRATADOS: ≥1 surto no último ano, durante o tratamento com uma droga modificadora do curso da doença (DMD), e evidência de ≥9 lesões hiperintensas em T2 ou ≥1 lesão captante de gadolínio, na ausência de toxicidade (intolerância, hipersensibilidade ou outro evento adverso) ou não adesão ao tratamento.12
O PCDT estabelece como critério para aprovação da terapia com natalizumabe a EMRR altamente ativa ou a EMRR de baixa ou moderada atividade com falha ou contraindicação ao uso de fingolimode.12
No sistema privado de saúde, o Anexo II da Resolução Normativa no 465 da Agência Nacional de Saúde Suplementar (ANS), de 2021, contém as diretrizes de utilização (DUTs) para cobertura de procedimentos pelos planos de saúde. O documento estabelece a obrigatoriedade de cobertura da terapia com natalizumabe, mesmo em primeira linha, para pacientes com EMRR grave e em rápida evolução, definida por ≥2 surtos incapacitantes em 1 ano e com ≥1 lesões realçadas por gadolínio em uma imagem de RMN cerebral ou aumento significativo das lesões em T2 comparativamente a uma RMN anterior recente.15
O documento da ANS estabelece também a obrigatoriedade de cobertura da terapia com natalizumabe em terceira ou quarta linha, após falha terapêutica das linhas prévias, que devem incluir o fingolimode. O alentuzumabe, o ocrelizumabe e o ofatumumabe têm cobertura obrigatória após falha do natalizumabe, entre outras condições.15
Terapia altamente efetiva: a melhor opção para pacientes com alta atividade da doença
A EM continua a ser uma condição incapacitante, mas temos hoje uma compreensão mais detalhada dos fatores genéticos e ambientais envolvidos em sua fisiopatologia. O diagnóstico precoce e acurado é crucial e baseia-se em critérios diagnósticos bem definidos, que incorporam anormalidades em exames de imagem e do LCR e devem ser aplicados àqueles que apresentam uma síndrome clinicamente isolada. O arsenal terapêutico disponível atualmente é amplo, sobretudo para pacientes com EMRR. A escolha da melhor opção terapêutica requer avaliação cuidadosa que pondere o perfil de efeitos colaterais e a eficácia clínica. Diversas DMDs foram desenvolvidas e aprovadas para pacientes com EMRR ou com CIS. A estratégia de escalonamento pode não ser efetiva para pacientes com alta atividade ou rápida progressão da doença, casos em que iniciar uma terapia altamente efetiva, como alentuzumabe ou natalizumabe, visando obter a remissão persistente da doença, pode ser uma estratégia mais adequada.16
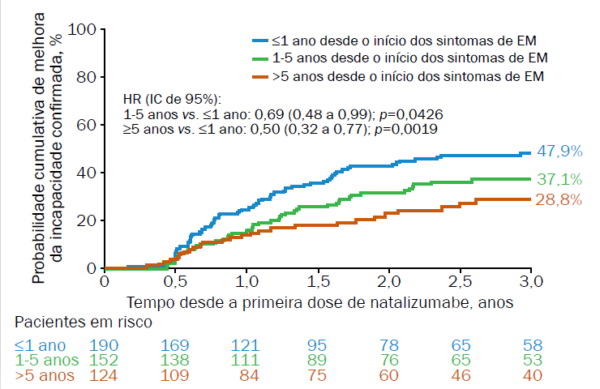
Dados do programa de observação de pacientes tratados com natalizumabe TOP (do original em inglês, Tysabri Observational Program) ilustram as diferenças na evolução da incapacidade funcional de pacientes com EM, por meio da Escala Expandida do Estado de incapacidade (EDSS), de acordo com a duração da doença no início do tratamento (Figuras 3 e 4).17
Figura 3. Alteração no escore EDSS após 3 anos de tratamento com natalizumabe, em relação ao escore basal, em pacientes virgens de tratamento que o iniciaram com natalizumabe ≤1 ano, 1 a 5 anos ou ≥5 anos após o início dos sintomas da EM
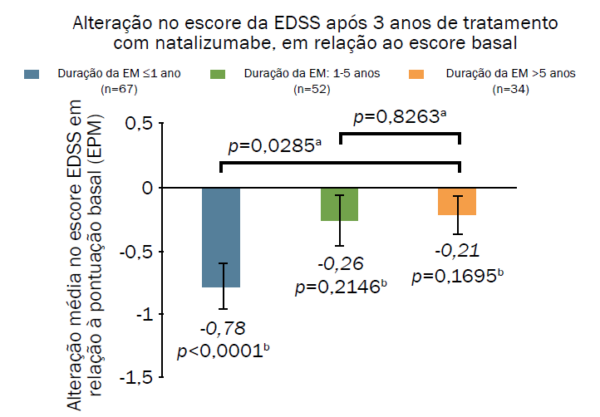
Figura 4. Probabilidade cumulativa de melhora confirmada da incapacidade após 3 anos de tratamento com natalizumabe, em relação ao escore basal, em pacientes com iniciaram o tratamento em ≤1 ano, 1 a 5 anos ou ≥5 anos após o início dos sintomas da EM
Clique aqui e descubra como Biogen Linc pode transformar sua rotina clínica.
Autoria

Felipe Resende Nobrega
Residência Médica em Neurologia (UNIRIO) • Mestre em Neurologia (UNIRIO) • Professor de Clínica Médica da UNESA
Prescreva medicação aos seus pacientes de forma gratuita e ilimitada
Como você avalia este conteúdo?
Sua opinião ajudará outros médicos a encontrar conteúdos mais relevantes.